|
本文介绍下载安装samtools及使用的方法,将对samtools的各种参数进行示例解析。
简介 samtools是一个用于操作sam和bam文件的工具合集。能够实现二进制查看、格式转换、排序及合并等功能,结合sam格式中的flag、tag等信息,还可以完成比对结果的统计汇总。同时利用linux中的grep、awk等操作命令,还可以大大扩展samtools的使用范围与功能。包含有许多命令。
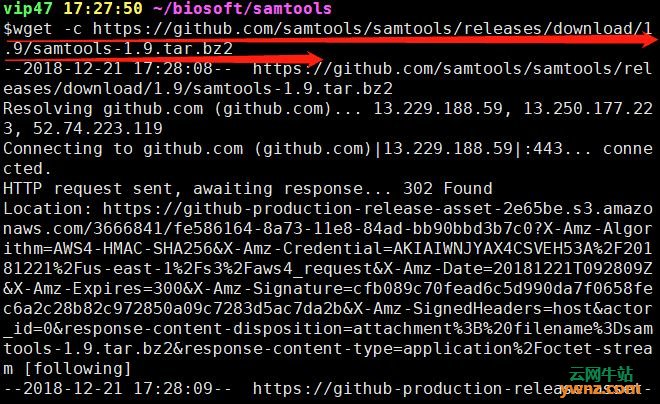
在linux系统中下载安装samtools的方法 如果已经装了conda(在linux系统中安装与卸载miniconda的方法),可以用conda直接安装,很方便。 这里我使用的最普通的安装方法: 首先,进入github中的samtools链接下载页面 找到最下面的安装包,然后右键,复制下载地址
打开linux,创建samtools文件夹,进入,然后用wget命令下载 mkdir ~/biosoft/samtools cd ~/biosoft/samtools wget -c https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2
下载完成后,可以看到当前目录下多了一个压缩文件samtools-1.9.tar.bz2
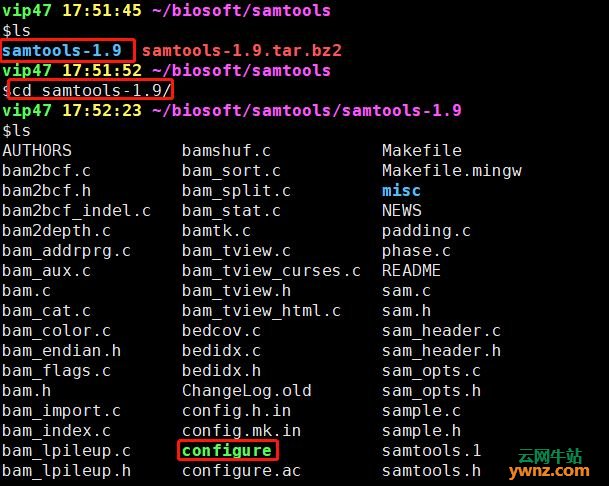
然后就是解压,观察发现,是bz压缩文件,所以加压的时候要用下面的解压方式 tar jxvf samtools-1.9.tar.bz2 解压之后,多了一个文件夹samtools-1.9,进入,发现里面有很多个文件,其中有一个configure的文件
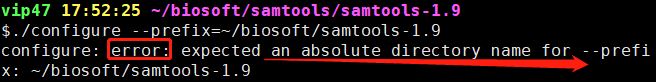
接下来就是安装了。用的是下面的命令 ./configure --prefix=~/biosoft/samtools-1.9 然后这里报错了
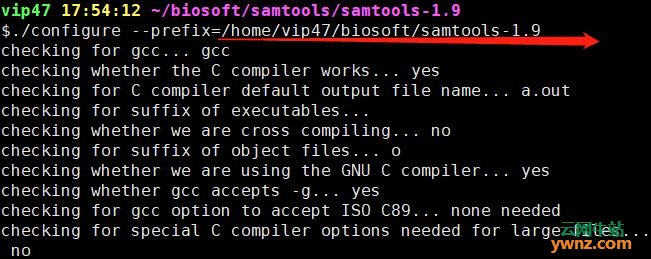
报错信息:需要输入一个绝对路径,哦,我刚才用的是相对路径。那就改一下路径 ./configure --prefix=/home/vip47/biosoft/samtools-1.9
ok,第一步安装成功,接下来就分别运行下面两行命令就搞定了。 make make install 调用的时候就用下面命令,如查看帮助文档 ./samtools --help
当然,我们要把它放到环境变量里面去,方法是 echo 'export PATH="/home/vip47/biosoft/samtools-1.9/bin:$PATH" ' >>~/.bashrc source ~/.bashrc 这样以后就可以随时随地的调用,不需要加路径。
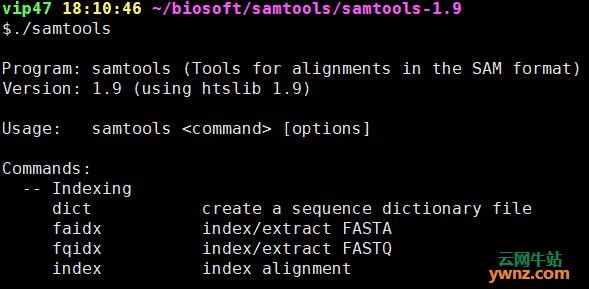
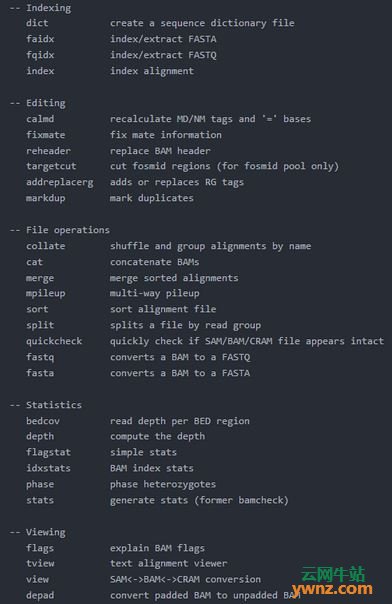
在linux系统中使用samtools的方法 先大致看一下samtools都有哪些命令 $samtools Program: samtools (Tools for alignments in the SAM format) Version: 1.9 (using htslib 1.9) Usage: samtools <command> [options] Commands如下图:
从上面我们可以看到,大致我5类命令块:Indexing,Editing,File operations,Statistics,Viewing,下面我们来看看几个常用的命令的使用方法示例。 1.view view命令的主要功能是:将sam文件与bam文件互换;然后对bam文件进行各种操作,比如数据的排序(sort)和提取(这些操作是对bam文件进行的,因而当输入为sam文件的时候,不能进行该操作);最后将排序或提取得到的数据输出为bam或sam(默认的)格式。 bam文件优点:bam文件为二进制文件,占用的磁盘空间比sam文本文件小;利用bam二进制文件的运算速度快。 view命令中,对sam文件头部(序列ID)的输入(-t或-T)和输出(-h)是单独的一些参数来控制的。 Usage: samtools view [options] | [region1 [...]] 下面的view命令的部分参数 默认情况下不加 region,则是输出所有的 region.options: -b output BAM # 该参数设置输出 BAM 格式,默认下输出是 SAM 格式文件 -h print header for the SAM output # 默认下输出的 sam 格式文件不带 header,该参数设定输出sam文件时带 header 信息 -H print SAM header only (no alignments) # 仅仅输出文件的头文件 -S input is SAM # 默认下输入是 BAM 文件,若是输入是 SAM 文件,则最好加该参数,否则有时候会报错。 -u uncompressed BAM output (force -b) # 该参数的使用需要有-b参数,能节约时间,但是需要更多磁盘空间。 -c print only the count of matching records # 仅输出匹配的统计记录 -L FILE only include reads overlapping this BED FILE [null] # 仅包括和bed文件存在overlap的reads -o FILE output file name [stdout] # 输出文件的名称 -F INT only include reads with none of the FLAGS in INT present [0] # 过滤flag,仅输出指定FLAG值的序列 -q INT only include reads with mapping quality >= INT [0] # 比对的最低质量值,一般认为20就为unique比对了,可以结合上述-bF参数使用使用提取特定的比对结果 -@ Number of additional threads to use [0] # 指使用的线程数 下面来看几个例子,如果想要比较直观的结果,可以用软件自带的测试数据,在example文件夹中 # 将sam文件转换成bam文件 samtools view -bS abc.sam > abc.bam # BAM转换为SAM samtools view -h -o out.sam out.bam # 提取比对到参考序列上的比对结果 samtools view -bF 4 abc.bam > abc.F.bam # 提取paired reads中两条reads都比对到参考序列上的比对结果,只需要把两个4+8的值12作为过滤参数即可 samtools view -bF 12 abc.bam > abc.F12.bam # 提取没有比对到参考序列上的比对结果 samtools view -bf 4 abc.bam > abc.f.bam # 提取bam文件中比对到caffold1上的比对结果,并保存到sam文件格式 samtools view abc.bam scaffold1 > scaffold1.sam # 提取scaffold1上能比对到30k到100k区域的比对结果 samtools view abc.bam scaffold1:30000-100000 $gt; scaffold1_30k-100k.sam # 根据fasta文件,将 header 加入到 sam 或 bam 文件中 samtools view -T genome.fasta -h scaffold1.sam > scaffold1.h.sam 2.sort sort对bam文件进行排序。 Usage: samtools sort [option] <in.bam> -o <out.prefix> -n Sort by read name #设定排序方式按short reads的ID排序。默认下是按序列在fasta文件中的顺序(即header)和序列从左往右的位点排序。 -m INT Set maximum memory per thread; suffix K/M/G recognized [768M] # 设置每个线程的最大内存,单位可以是K/M/G,默认是 768M。对于处理大数据时,如果内存够用,则设置大点的值,以节约时间。 -t TAG Sort by value of TAG. Uses position as secondary index (or read name if -n is set) # 按照TAG值排序 -o FILE Write final output to FILE rather than standard output # 输出到文件中,加文件名 例子: # tmp.bam 按照序列位置排序,并将结果输出到tmp.sort.bam samtools sort -n tmp.bam -o tmp.sort.bam samtools view tmp.sort.bam 3.merge和cat merge将多个已经sort了的bam文件融合成一个bam文件。融合后的文件不需要则是已经sort过了的。而cat命令不需要将bam文件进行sort。 Usage: samtools merge [-nurlf] [-h inh.sam] [-b <bamlist.fofn>] <out.bam> <in1.bam> [<in2.bam> ... <inN.bam>] Options: -n Input files are sorted by read name # 输入文件是经过sort -n的 -t TAG Input files are sorted by TAG value # 输入文件是经过sort -t的 -r Attach RG tag (inferred from file names) # 添加上RG标签 -u Uncompressed BAM output # 输出未压缩的bam -f Overwrite the output BAM if exist # 覆盖已经存在的bam -1 Compress level 1 # 1倍压缩 -l INT Compression level, from 0 to 9 [-1] # 指定压缩倍数 -R STR Merge file in the specified region STR [all] -h FILE Copy the header in FILE to <out.bam> [in1.bam] $samtools cat Usage: samtools cat [options] <in1.bam> [... <inN.bam>] samtools cat [options] <in1.cram> [... <inN.cram>] Options: -b FILE list of input BAM/CRAM file names, one per line -h FILE copy the header from FILE [default is 1st input file] -o FILE output BAM/CRAM 4.index 对排序后的序列建立索引,并输出为bai文件,用于快速随机处理。在很多情况下,特别是需要显示比对序列的时候,bai文件是必不可少的,例如之后的tview命令。 Usage: samtools index <in.bam> [out.index] samtools index abc.sort.bam 5.faidx 对fasta文件建立索引,生成的索引文件以.fai后缀结尾。该命令也能依据索引文件快速提取fasta文件中的某一条(子)序列 Usage: samtools faidx <file.fa|file.fa.gz> [<reg> [...]]samtools faidx <file.fa|file.fa.gz> [<reg> [...]] 如对基因组文件建立索引 samtools faidx genome.fasta # 生成了索引文件genome.fasta.fai,是一个文本文件,分成了5列。第一列是子序列的名称;第二列是子序列的长度;个人认为“第三列是序列所在的位置”,因为该数字从上往下逐渐变大,最后的数字是genome.fasta文件的大小;第4和5列不知是啥意思。于是通过此文件,可以定位子序列在fasta文件在磁盘上的存放位置,直接快速调出子序列。 # 由于有索引文件,可以使用以下命令很快从基因组中提取到fasta格式的子序列 samtools faidx genome.fasta scffold_10 > scaffold_10.fasta 6.tview tview能直观的显示出reads比对基因组的情况,和基因组浏览器有点类似。可视化一般用IGV比较好,不建议用tview Usage: samtools tview <aln.bam> [ref.fasta] 当给出参考基因组的时候,会在第一排显示参考基因组的序列,否则,第一排全用N表示。 按下 g,则提示输入要到达基因组的某一个位点。例子“scaffold_10:1000"表示到达第10号scaffold的第1000个碱基位点处。 使用H(左)J(上)K(下)L(右)移动显示界面。大写字母移动快,小写字母移动慢。 使用空格建向左快速移动(和 L 类似),使用Backspace键向左快速移动(和 H 类似)。 Ctrl+H 向左移动1kb碱基距离; Ctrl+L 向右移动1kb碱基距离 可以用颜色标注比对质量,碱基质量,核苷酸等。30~40的碱基质量或比对质量使用白色表示; 20~30黄色;10~20绿色;0~10蓝色。 使用点号‘.‘切换显示碱基和点号;使用r切换显示read name等 还有很多其它的使用说明,具体按 ? 键来查看。 7.flagstat 给出BAM文件的比对结果,并输出比对统计结果。除了-@参数指定线程,没有其他的参数 Usage: samtools flagstat [options] <in.bam> samtools flagstat tmp.bam 20000 + 0 in total (QC-passed reads + QC-failed reads) # 总共的reads数 0 + 0 secondary 0 + 0 supplementary 0 + 0 duplicates 18995 + 0 mapped (94.98% : N/A) # 总体上reads的匹配率 20000 + 0 paired in sequencing # 有多少reads是属于paired reads 10000 + 0 read1 # reads1中的reads数 10000 + 0 read2 # reads2中的reads数 18332 + 0 properly paired (91.66% : N/A) # 完美匹配的reads数和比例:比对到同一条参考序列,并且两条reads之间的距离符合设置的阈值 18416 + 0 with itself and mate mapped # paired reads中两条都比对到参考序列上的reads数 579 + 0 singletons (2.90% : N/A) # 单独一条匹配到参考序列上的reads数,和上一个相加,则是总的匹配上的reads数。 0 + 0 with mate mapped to a different chr # paired reads中两条分别比对到两条不同的参考序列的reads数 0 + 0 with mate mapped to a different chr (mapQ>=5) # 同上一个,只是其中比对质量>=5的reads的数量 8.depth 得到每个碱基位点的测序深度,并输出到标准输出。输入的bam文件必须先做samtools index Usage: samtools depth [-r reg] [-q baseQthres] [-Q mapQthres] [-b in.bed] <in1.bam> [...] -r <chr:from-to> region # 后面跟染色体号(region) -a output all positions (including zero depth) # 输入所有位置的序列,包括测序深度为0的 -q <int> base quality threshold [0] # 碱基质量阈值 -Q <int> mapping quality threshold [0] # 比对的质量阈值 举例: samtools depth tmp.index.bam > tmp.depth.bam 9.其它有用的命令 reheader 替换bam文件的头 $ samtools reheader <in.header.sam> <in.bam> idxstats 统计一个表格,4列,分别为”序列名,序列长度,比对上的reads数,unmapped reads number”。第4列应该是paired reads中有一端能匹配到该scaffold上,而另外一端不匹配到任何scaffolds上的reads数。 samtools idxstats <aln.bam> 10.将bam文件转换为fastq文件 有时候,我们需要提取出比对到一段参考序列的reads,进行小范围的分析,以利于debug等。这时需要将bam或sam文件转换为fastq格式。 wget http://www.hudsonalpha.org/gsl/static/software/bam2fastq-1.1.0.tgz tar zxf bam2fastq-1.1.0.tgz cd bam2fastq-1.1.0 make ./bam2fastq <in.bam> 11.mpileup samtools还有个非常重要的命令mpileup,以前为pileup。该命令用于生成bcf文件,再使用bcftools进行SNP和Indel的分析。bcftools是samtool中附带的软件,在samtools的安装文件夹中可以找到。 用法: Usage: samtools mpileup [-EBug] [-C capQcoef] [-r reg] [-f in.fa] [-l list] [-M capMapQ] [-Q minBaseQ] [-q minMapQ] in.bam [in2.bam [...]] 最常用的参数有2: -f 来输入有索引文件的fasta参考序列; -g 输出到bcf格式。用法和最简单的例子如下 samtools mpileup -f genome.fasta abc.bam > abc.txt samtools mpileup -gSDf genome.fasta abc.bam > abc.bcf samtools mpileup -guSDf genome.fasta abc.bam | bcftools view -cvNg - > abc.vcf mpileup不使用-u或-g参数时,则不生成二进制的bcf文件,而生成一个文本文件(输出到标准输出)。该文本文件统计了参考序列中每个碱基位点的比对情况;该文件每一行代表了参考序列中某一个碱基位点的比对结果。比如:
mpileup生成的结果包含6行:参考序列名;位置;参考碱基;比对上的reads数;比对情况;比对上的碱基的质量。其中第5列比较复杂,解释如下: ‘.’代表与参考序列正链匹配。 ‘,’代表与参考序列负链匹配。 ‘ATCGN’代表在正链上的不匹配。 ‘atcgn’代表在负链上的不匹配。 ‘*’代表模糊碱基 ‘’代表匹配的碱基是一个read的开始;’‘后面紧跟的ascii码减去33代表比对质量;这两个符号修饰的是后面的碱基,其后紧跟的碱基(.,ATCGatcgNn)代表该read的第一个碱基。 ‘$’代表一个read的结束,该符号修饰的是其前面的碱基。 正则式’+[0-9]+[ACGTNacgtn]+’代表在该位点后插入的碱基;比如上例中在scaffold_1的2847后插入了9个长度的碱基acggtgaag。表明此处极可能是indel。 正则式’-[0-9]+[ACGTNacgtn]+’代表在该位点后缺失的碱基; 12.samtools rmdup NGS上机测序前需要进行PCR一步,使一个模板扩增出一簇,从而在上机测序的时候表现出为1个点,即一个reads。若一个模板扩增出了多簇,结 果得到了多个reads,这些reads的坐标(coordinates)是相近的。在进行了reads比对后需要将这些由PCR duplicates获得的reads去掉,并只保留最高比对质量的read。使用rmdup命令即可完成. Usage: samtools rmdup [-sS] -s rmdup for SE reads # 对single-end reads。默认情况下,只对paired-end reads -S treat PE reads as SE in rmdup (force -s) # 将Paired-end reads作为single-end reads处理。 至此,samtools的基本使用方法已经讲完了。
相关主题 |